

Regulatory Aspects of
Paediatric Drug Development.
by Dr Leticia Monjas Gómez
INTRODUCTION
Paediatric drug development is a complex and challenging process, due to ethical, methodological, operational and financial limitations. Paediatrics are not a uniform sub-group and the medical needs, biological and physiological characteristics of neonates, for example, are very different to that of adolescents. Therefore, additional research is often needed for the different subpopulations, making the process of developing paediatric medicines more complex.
Paediatric trials are still not widely accepted by society for fear of exposing children to uncertain treatment effects. Because of the small patient pool, paediatric studies have high infrastructure needs, requiring trials to take place across multiple sites and countries.
In the current climate, there is broad consensus that children deserve access to medicines that have been specifically developed and researched for this population. Until recently, however, many products prescribed and administered to children were based on physicians’ own experience, rather than on the results of clinical research. Moreover, medicines were often not available in a pharmaceutical form that was suitable for children. Paediatricians had to rely on medicines authorised for adults by adapting the dosage and form.
This off-label use of adult medicines comes with the risk of inefficacy and/or adverse reactions in children. Side effects that may not affect adults can be serious in children.
The Paediatric Medicine Regulation (PMR) in the European Union (EU) and its equivalent legislation in the United States (US) were adopted to address this problem, and have stimulated new research efforts for the paediatric population. Before the PMR introduction in Europe, only one third of approved drugs had a paediatric indication listed in their marketing authorisations. Ten years later, that had risen to 70% for all newly approved medicines (State of Paediatric Medicines in the EU – 10 years of the EU Paediatric Regulation).
Specific legislation in the EU and the US provides incentives and obligations or requirements, under certain circumstances, for development of paediatric medicinal products. These programmes are implemented by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). Incentives and obligations are similar but not identical in the two regions. The key regulations, guidelines and publications related to paediatric drug development in the EU and the US are listed in Table 1.

Table 1: Paediatric Drug Development Regulations and Guidelines
EU
The European Paediatric Regulation (EC) No 1901/2006 came into effect on 26 January 2007, with the aim of assuring that children from birth to less than 18 years have safe access to medicinal products. The paediatric regulation established the Paediatric Committee (PDCO), which is the scientific committee responsible for activities on medicines for children in the EU.
The PDCO support the development of such medicines by providing scientific expertise and defining paediatric needs. The paediatric regulation made a paediatric investigation plan (PIP) mandatory. A PIP is a development plan aimed at ensuring that the necessary data are obtained through studies in children, to support the authorisation of a medicine for children. All marketing authorisation applications (MAA) for new medicines have to include the results of studies as described in an agreed PIP, unless the medicine is exempt because of a deferral or waiver. This requirement also applies when a marketing-authorisation holder wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorised and covered by intellectual property rights. These requirements also apply to orphan-designated products. However, generic, hybrid, or biosimilar biological products are exempt, as well as homeopathic and traditional herbal products, and those to be authorised using the “well-established use” legal basis.
Incentives are available for sponsors, such as a 6-month extension of the supplementary protection certificate (SPC) and additional 2 years of market exclusivity for paediatric orphan medicines. In addition, the EMA offers free scientific advice on questions relating to paediatric development.
Medicines not covered by an SPC or eligible for an SPC, developed specifically for paediatric use and with an age-appropriate formulation, can benefit from a paediatric-use marketing authorisation (PUMA), which offers 10 years of market protection as an incentive.
PIP ASSESSMENT PROCEDURE
According to Article 16 of the paediatric regulation (Regulation (EC) No 1901/2006), applications should be submitted, unless duly justified, ‘not later than upon completion of the human pharmaco-kinetic (PK) studies in adults’, as specified in Section 5.2.3 of Part 1 of Annex 1 of Directive 2001/83/EC. The timing of submission should not be later than the end of healthy subject or patient PK, which can coincide with the initial tolerability studies, or the initiation of the adult phase 2 studies (proof-of-concept studies); it should not be after initiation of pivotal trials or confirmatory (phase 3) trials. Applicants are welcome to submit their PIP applications during or even before initial PK studies in adults. Submitting a PIP application for a new active substance during confirmatory or phase 3 trials in adults, or after starting clinical trials in children, is likely to be considered unjustified. A PIP is assessed by the PDCO and follows a set procedure with defined timelines (Figure 1).

Figure 1: PIP Assessment Procedure
The evaluation process takes the form of a 120-day procedure. The Day 30 summary report is the first communication from the PDCO, which consists of review and feedback from the paediatric coordinator, rapporteur and peer-reviewer. This document also contains a summary of the PDCO discussion regarding the application. This report provides the applicant with an initial indication of the position of the PDCO, and what the potential requests for modification (RfM) may be in the Day 60 report.
The Day 60 document details the official list of outstanding issues/RfM that the PDCO is requesting from the applicant in order to consider a positive outcome for the application. There is a pause (clock stop) at Day 60, which is usually three months in duration, although this can be shortened or lengthened depending on the procedure. Once the applicant’s responses have been submitted, the clock begins again at Day 61.
The Day 90 summary report follows, which includes feedback from the paediatric coordinator, rapporteur, peer-reviewer, and a summary of the PDCO discussion regarding the updated application. If any questions remain outstanding after the third discussion with the PDCO, then the PDCO or the applicant can request an oral explanation. This enables the applicant to speak directly to the whole Committee.
The final outcome of the PDCO discussion is available at Day 120, when the PDCO opinion and summary report are provided to the applicant.
WAIVERS
The PDCO may grant PIP waivers exempting the sponsor from submitting a PIP. There are three legal grounds for PIP waivers:
The specific medicinal product or class of medicinal products is likely to be ineffective or unsafe in part or all of the paediatric population;
The disease or condition for which the medicine(s) is intended occurs only in adults (or only in some paediatric subsets);
There is a lack of ‘significant therapeutic benefit’ over existing treatments for paediatric patients, or there is a justification that there are feasibility issues meaning that significant therapeutic benefit cannot be demonstrated (e.g., for exceedingly rare conditions in paediatric patients when informative studies cannot be performed, or when clinical studies cannot be conducted).
Waivers can be granted for specific products, or for whole classes of medicinal products. There are three types of waiver:
Class waiver – according to a list issued by the PDCO of conditions that only occur in adults
Full waiver – for all paediatric subset(s) and indication(s)
Partial waiver – for one or more paediatric subset(s) and indication(s).
A waiver may be reviewed and changed. If a waiver is revoked, the requirement to submit data according to an agreed PIP will not apply for 36 months.
DEFERRALS
Deferrals can be granted for some or all of the paediatric studies included in an agreed PIP. This means that these studies can be initiated and/or completed after applying for marketing authorisation for adults, for the same condition. Deferrals allow an applicant to delay development of the medicine in children and can be justified on scientific and technical grounds, or on grounds related to public health. In any event, a deferral shall be granted when it is appropriate to conduct studies in adults prior to initiating studies in the paediatric population or when studies in the paediatric population will take longer to conduct than studies in adults. An opinion in favour of a deferral shall specify the time limits for initiating or completing the measures concerned.
COMPLIANCE CHECK
Applicants can request that a PIP compliance check (CC) is carried out before submitting a MAA. Alternatively, the CC will be carried out as part of the validation of the application. Applicants are strongly recommended to apply for the CC before submission of the MAA to avoid delays during the validation phase. This check verifies that all the measures mentioned in the PIP decision, including the timelines for the conduct of the studies or collection of the data, have been completed in accordance with the key elements specified in the PIP decision.
MODIFICATIONS OF AN AGREED PIP
According to Art 22 of the Paediatric Regulation (Regulation (EC) No 1901/2006), the applicant may request a modification of an agreed PIP if they encounter difficulties with its implementation as to render the plan unworkable or no longer appropriate. Applications to modify the PIP are particularly important if new information may have an impact on the nature or timelines for completion of one of the key elements. The modification procedure takes 60 days or less.
SCIENTIFIC ADVICE
Applicants can request scientific advice from EMA in preparation of a PIP, which is free of charge for questions relating to the development of paediatric medicines. Advice can also follow the submission of a PIP, for example seeking advice on combined adult and paediatric development in light of the PIP requirements.
EUROPEAN NETWORK OF PAEDIATRIC AT THE EMA
The European Network of Paediatric Research at the EMA (Enpr-EMA) is a network of research networks, investigators and centres with recognised expertise in performing clinical studies in children.
Enpr-EMA enables networking and collaboration with members from within and outside the EU, including academia and the pharmaceutical industry. It acts as a platform for sharing good practices as well as a pan-European voice for promoting research into medicines for children.
The network does not perform clinical trials or fund studies or research or decide on areas for paediatric research, as this is the responsibility of Member States, the European Commission or each individual member organisation.
PLAN TO BOOST THE DEVELOPMENT OF MEDICINES FOR CHILDREN
There are still a number of challenges to be addressed, for example developing medicines for diseases that only affect children, or for diseases that manifest differently in adults and children. The European Commission published in 2017 the ten-year report on implementation of the Paediatric Regulation, which identified actions in five key areas to address these challenges:
Identifying paediatric medical needs;
Strengthening co-operation between decision makers;
Ensuring timely completion of PIPs;
Improving the handling of PIP applications; and
Increasing transparency around paediatric medicines.
It is expected that the implementation of the proposed actions will increase the efficiency of paediatric regulatory processes in the current legal framework and boost the availability of medicines for children.
UK
From 1 January 2021, the Medicines and Healthcare Products Regulatory Agency (MHRA) is the UK’s standalone medicines and medical devices regulator. The legal requirements for UK PIPs are set out in the Human Medicines Regulations 2012, as amended by the Human Medicines Regulations (Amendment etc.) (EU Exit) Regulations 2019 (HMRs).
The PIP application process for applicants is simplified by offering an expedited assessment where possible, and by mirroring the submission format, content and terminology of the EU-PIP system. The MHRA is taking decisions on PIP and waiver opinions, modifications and compliance statements to support paediatric market authorisation decisions, while acknowledging that Northern Ireland continues to be part of the EU’s system for paediatric medicines development including agreement of EU PIPs or waivers.
Applicants should include information relevant to the UK, particularly with respect to any areas of unmet therapeutic need that their product intends to cover. Parallel submission of PIPs to EMA and MHRA is strongly supported to allow robust parallel assessment and finally alignment of the agreed paediatric plans across jurisdictions.
UNMET NEEDS IN THE UK PAEDIATRIC POPULATION
The unmet needs for the UK paediatric population are defined by:
Therapeutic areas identified by UK health bodies as high priority public health concerns
Product development in conditions identified after consultations with UK experts and patient groups, including those for rare diseases identified under the auspices of the Department of Health and Social Care (DHSC) policy paper - UK strategy for rare diseases
Product development in conditions (or paediatric groups) identified as critically important in the Paediatric Regulation 10 year report
Products which are intended to be authorised as orphan medicines
Products that fulfil the criteria of promising/innovative new products and are part of an accelerated MHRA submission or assessment pathway.
PIP SUBMISSIONS
EU-PIPs, modifications and full product specific waivers with an EMA decision agreed or with a positive opinion by the PDCO before 1 January 2021, are adopted as UK-PIPs on or after that date, therefore, these PIP applications do not require re-submission to the MHRA. If the PDCO has issued a negative opinion, the MHRA treats the application as refused. However, applicants can submit an updated PIP to the MHRA which addresses the reasons for refusal.
For UK PIP submissions made after 1 January 2021 to the MHRA, information should be provided on whether there is:
An agreed EU-PIP and the opinion and supporting documentation is included
An ongoing EU-PIP assessment and its timeline in the PDCO assessment cycle
Any current scientific divergence between the submitted PIP application and the EU-PIP
If there is no corresponding EU PIP submission or if the PDCO opinion is negative, a full assessment of the UK-PIP is required. The assessment criteria are based on national paediatric public health needs:
Unmet UK paediatric needs
Paediatric only development particularly for an innovative product
The incidence of the disease in the UK population
The relevance of the scientific arguments by EMA / PDCO in the summary report (SR) to the UK paediatric population
Any additional safety or efficacy concerns for the UK population
The nature and number of licensed products already available for the intended paediatric indication
The feasibility of performing the proposed paediatric studies in the UK only
PIP is to support a UK Paediatric Use Marketing Authorisation (PUMA)
Therefore, the assessment pathways for UK PIP submissions depend on the status of the EU PIP (Figure 2).
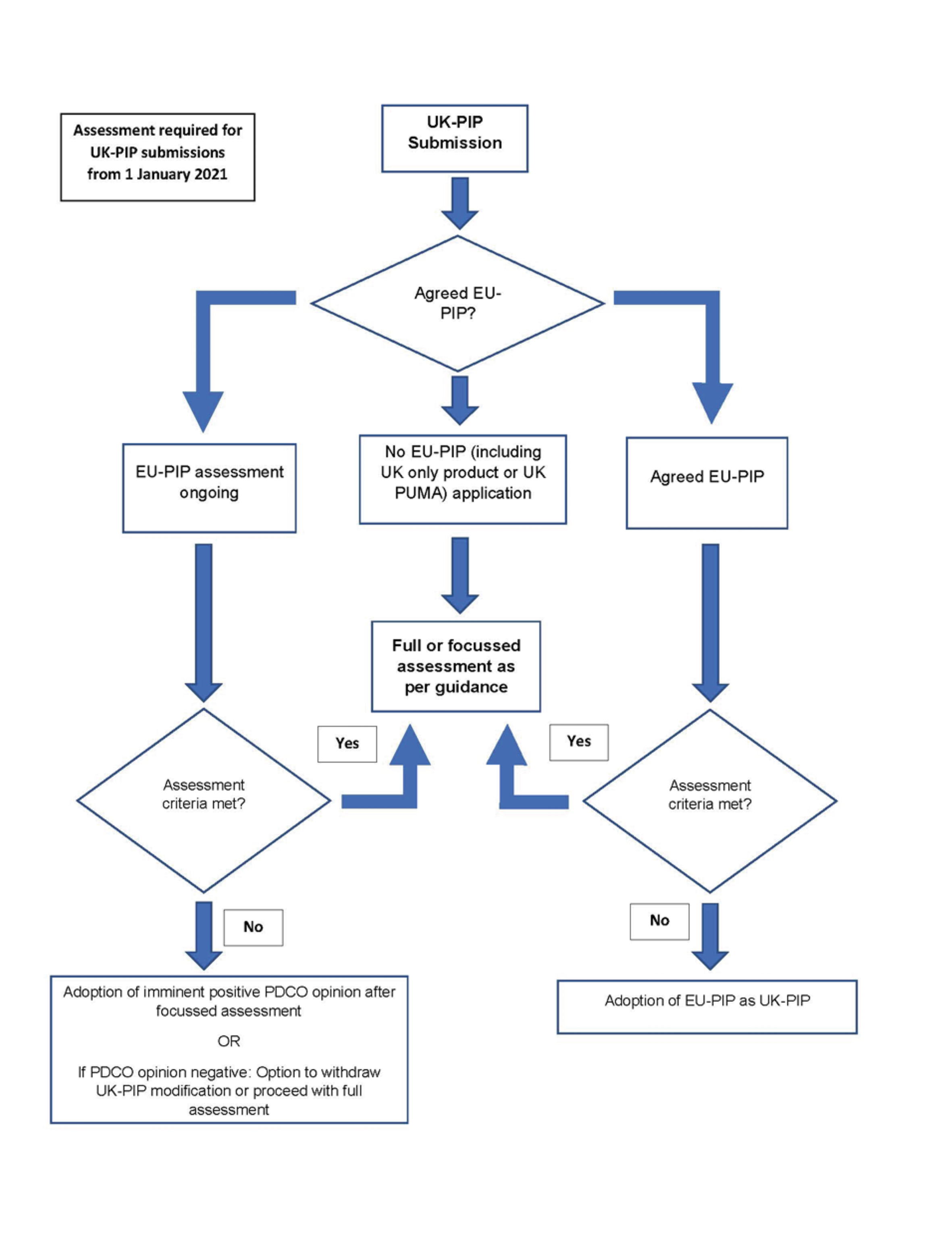
Figure 2: Assessment Pathways for UK-PIPs from 1 January 2021
CLASS WAIVERS
The current EMA class waivers list was adopted by the MHRA from 1 January 2021. The MHRA aims to accept a positive EMA opinion on a class waiver request. Where there is no EMA opinion, or a negative EMA opinion, a MHRA assessment is undertaken.
DEFERRALS
In accordance with Regulation 50C of the HMRs, a deferral may be granted when it is appropriate to conduct studies in adults prior to initiating studies in the paediatric population; or studies in the paediatric population will take longer to conduct than studies in adults.
COMPLIANCE CHECK
A positive PDCO CC or interim CC is adopted as the UK CC outcome unless subsequent modifications have led to divergence between the UK- and EU-PIPs. However, the applicant must pay particular attention to the agreed timelines of those measures which would need to be completed after the PDCO CC to ensure compliance on the date of the UK MA submission. The PDCO compliance outcome documents should be submitted ahead of, or at the time of MAA. At the end of the CC procedure an MHRA compliance report will be issued to the applicant.
US
In the US, since 1997, there have been legislative and regulatory approaches to address paediatric product development for children from birth to less than 17 years [21 CFR 201.57(c)(9)(iv)]. The current legislation is the Best Pharmaceuticals for Children Act (BPCA), which provides incentives but is voluntary; and the Paediatric Research Equity Act (PREA), which establishes requirements to perform paediatric development under certain circumstances but does not offer incentives.
Both acts were made permanent in 2012, with modifications, in Title V of the FDA Safety and Innovation Act (FDASIA). To meet US paediatric research obligations, one must address PREA requirements. However, to obtain paediatric exclusivity, the BPCA process, which requires FDA to issue a Written Request (WR), must also be followed. To obtain exclusivity in the US, the sponsor needs to voluntarily perform the studies in the WR.
The FDA’s requirement for sponsors to submit an initial paediatric study plan (iPSP) are described in section 505B(e) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). A sponsor who is planning to submit a marketing application (or supplement to an application) for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration is required to submit an iPSP unless the drug is for an indication for which orphan designation has been granted. However, it should be noted that as of 18 August 2020, a sponsor who is planning to submit an original application for a new active ingredient that is subject to the molecularly targeted cancer drug provision of the PREA (i.e., the drug that is the subject of the application is intended for the treatment of an adult cancer and is directed at a molecular target that the FDA determines to be substantially relevant to the growth or progression of a paediatric cancer) is also required to submit an iPSP, regardless of whether the drug is for an indication for which orphan designation has been granted. By statute, a biosimilar product that has not been determined to be interchangeable with the reference product is considered to have a new active ingredient for purposes of PREA.
The sponsor must submit an iPSP for any new application or supplement that is subject to PREA, regardless of whether the FDA has previously granted waivers or deferrals under PREA for the same drug. Additionally, for drugs that are being developed specifically for use in paediatric populations, the sponsor should submit an iPSP.
IPSP ASSESSMENT PROCEDURE
A sponsor must submit an iPSP, if required under PREA, before the date on which the sponsor submits the required assessments or investigation and no later than either 60 calendar days after the date of the end-of-phase 2 meeting or such other time as agreed upon between FDA and the sponsor.
In the absence of an end-of-phase 2 meeting, the sponsor should submit the iPSP as early as practicable but before the initiation of any phase 3 studies, or any combined phase 2 and phase 3 studies, of the drug that is the subject of the iPSP.
If a phase 3 study, or a combined phase 2 and phase 3 study, will not be conducted or will be conducted but not under investigational new drug (IND), the sponsor should submit the iPSP no later than 210 calendar days before it submits a marketing application or supplement.
The sponsor should submit the iPSP to the relevant drug’s IND for review by the Centre for Drug
Evaluation and Research (CDER) or Centre for Biologics Evaluation and Research (CBER) as appropriate. In cases where the sponsor has no active IND for the drug, but the sponsor expects to open the IND with an initial phase 3 study, the sponsor should submit the iPSP as a pre-IND submission. Where the drug development programme includes the possibility of using expedited programs, the FDA encourages the sponsor to have discussions about the paediatric development plans with the review division as early as possible.
After the sponsor submits an iPSP, the FDA has 90 days to review and provide a written response, or meet with the sponsor to discuss the iPSP, as appropriate. This review process includes consultation with FDA’s internal Paediatric Review Committee (PeRC). The sponsor then has a second 90-day period during which it may review FDA comments and initiate any negotiations to discuss the iPSP. By the end of this second 90-day review period, the sponsor must submit an agreed iPSP. The FDA has 30 days after receipt of the agreed iPSP to review and issue correspondence confirming agreement or stating disagreement. If the FDA does not agree, the iPSP is considered a non-agreed iPSP. The total length of time for review of an iPSP should not exceed 210 days (Figure 3). A sponsor should not submit an application for marketing, or supplement, until the FDA confirms agreement on the iPSP.

Figure 3: iPSP assessment procedure
WAIVERS
Under PREA, sponsors may request a waiver of paediatric assessments, or reports on the molecularly targeted paediatric cancer investigation, at the time of the submission of the new drug application (NDA), biologics license application (BLA), or supplement. FDA does not formally grant or deny a request for a waiver in response to the iPSP. The FDA formally grants a waiver(s) when it issues an approval letter for an NDA, BLA, or supplement. PREA authorises the FDA to grant a full waiver of required paediatric assessments or reports on the molecularly targeted paediatric cancer investigation if it finds that:
Necessary studies are impossible or highly impracticable (if, for example, the number of patients is small or the patients are geographically dispersed)
There is evidence strongly suggesting that the drug would be ineffective or unsafe in all paediatric age groups
The drug does not represent a meaningful therapeutic benefit over existing therapies for paediatric patients and is not likely to be used in a substantial number of paediatric patients
In addition, PREA authorises the FDA to grant a partial (i.e., with respect to a specific paediatric age group) waiver of required paediatric assessments or reports on the molecularly targeted paediatric cancer investigation if it finds that any of the three points above apply to a subset of the paediatric population, or the applicant can demonstrate that reasonable attempts to produce a paediatric formulation necessary for that age group have failed.
For indications that have extremely limited applicability to the paediatric population because the pathophysiology of the relevant disease occurs for the most part only in adults, the FDA generally does not intend to require sponsors to provide additional evidence that studies are impossible or highly impracticable.
DEFERRALS
Under PREA, sponsors may request deferral of paediatric assessments or reports on the molecularly targeted paediatric cancer investigation at the time of the submission of the NDA, BLA, or supplement.
The FDA formally grants a deferral when it issues an approval letter for an NDA, BLA, or supplement. It is important to include in the iPSP any plan to submit a request for a deferral for any study required under PREA that will not be submitted as part of a planned application.
At the time of approval of an application, the FDA may grant a deferral of required paediatric assessments or reports on the molecularly targeted paediatric cancer investigation if it finds that:
The drug is ready for approval for use in adults before paediatric studies are complete
Paediatric studies should be delayed until additional safety or effectiveness data have been collected
There is another appropriate reason for deferral.
AMENDMENTS TO AN AGREED PSP
Sponsors can request an amendment to an agreed PSP at any time. Once the FDA accepts for filing an application or supplemental application, it is not necessary to submit amendments to the PSP because changes to the plan for paediatric development can be negotiated during the review cycle as appropriate. The timeline for submission, review, and agreement on an amended PSP is the same as on an iPSP. If the FDA does not agree to the amended PSP, the original agreed PSP remains in force.
PROPOSED PAEDIATRIC STUDY REQUESTS
As noted above, exclusivity is a separate legislation and process for the US. As such, the proposed paediatric study requests (PPSR) is a separate document from the PSP. It does not serve as a substitute for the PSP. The PPSR and the PSP serve distinct purposes as they address separate paediatric legislations. BPCA provides a financial incentive to companies in the form of additional marketing exclusivity. In cases where conducting clinical studies in children is not obligatory, sponsors are able to submit proposed paediatric study requests (PPSRs) to the FDA, in accordance with Section 505(A) of The Modernization Act. Using this approach, sponsors are able to take advantage of the 6-month extra market exclusivity which occurs upon completion of paediatric studies. Notably, the PPSR will only be granted if the sponsor completes the FDA requirements in the issued Written Response.
To obtain Written Requests for paediatric studies, sponsors should submit the PPSR to the appropriate new drug review division(s) of FDA, indicating the studies necessary to use the active moiety appropriately in paediatric subpopulations.
Written Requests from FDA outline certain studies which should be performed to determine if the use of a drug may have meaningful health benefits in the paediatric population. Issuance of a WR does not require the sponsor to conduct the suggested paediatric studies, and the final decision to perform the paediatric studies lies with the sponsor. Sponsors should plan submissions of PPSRs with sufficient time to enable the FDA to review the PPSR, engage in dialogue with the sponsor if required, issue a WR, and ensure initiation, completion and filing of study reports before expiration of a patent or exclusivity period. It is estimated that the FDA may take approximately 120 days after submission of a PPSR to provide a response. Written Requests can also be amended, either initiated by FDA or at the request of the sponsor. If initiated by the sponsor, they must obtain an amended WR from FDA prior to the conduct of the revised studies.
JOINT EMA/FDA PROCEDURES
In 2007, the Paediatric Cluster was established as a forum for informal exchange of scientific information. This Cluster includes the FDA, EMA, and regulators from Japan, Canada, and Australia. This co-operation is constantly evolving and new initiatives from EMA and FDA for a harmonised approach on paediatric development plans make the common goal of a single, aligned paediatric plan feasible. The EMA and the FDA published joint guidance in March 2021 for medicine developers submitting a PIP to EMA and an iPSP to the FDA for the use of cancer medicines in children. The purpose is to expedite development and authorisation of such medicines, given the rarity of childhood cancers. For certain therapeutic areas, Agencies can also provide special provisions for paediatric development. For example, EMA and the FDA published a joint procedural information document in June 2020 for COVID-19 vaccines or treatments. The aim of the joint document was to facilitate the simultaneous submission of paediatric development plans.
SUMMARY
The paediatric regulations in Europe and the US are aligned on common scientific principles and are designed to incentivise the development of medicinal products for paediatric patients.
Although there are many similarities between their legislations and guidelines, there are also a number of differences, particularly regarding the exclusivity process, exemptions, expected timelines for submission and assessment, and the evaluation process (Table 2).

*Regulatory aspects of the EU apply to the UK [Human Medicines Regulations 2012, as amended by the Human Medicines Regulations (Amendment etc.)
(EU Exit) Regulations 2019 (HMRs)].
Abbreviations: BPCA: Best Pharmaceuticals for Children Act, CHMP: Committee for Medicinal Products for Human Use, EoP: end-of-phase, FDASIA: FDA Safety and Innovation Act, MAA: marketing authorisation application, PDCO: paediatric committee, PeRC: paediatric review committee, PIP: paediatric investigation plan, PREA: Paediatric Research Equity Act, PSP: paediatric study plan, SPC: supplementary protection certificate, WR: written request.