

Making The Most of Opportunities to Interact With European Regulators.
by Harriet Thomasson
Introduction
In order for a new drug to reach the market in the EU, it must meet three key criteria: Quality, Safety and Efficacy, as defined by the regulatory authorities. It is common knowledge that the process of drug development is vastly expensive, and it is crucial for pharmaceutical companies to ensure that budgets are spent as efficiently as possible to develop a data package that demonstrates fulfilment of the Quality, Safety and Efficacy criteria.
Although there are many published guidelines and regulations available to inform drug development, there is no substitute for interacting directly with a regulatory agency to agree the data requirements for a specific development programme. To facilitate this dialogue, the European Medicines Agency (EMA) provides a formal process for this interaction through Scientific Advice (Article 57-1 (n) of Regulation (EC) No 726/2004). Developers of orphan medicines are also eligible for a similar procedure, Protocol Assistance (under Article 6 of the Regulation on Orphan Medicinal Products (EC) 141/2000).
Alternatively, a developer may choose to interact with any number of European National Competent Authorities (NCAs), such as the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA), or Germany’s Federal Institute for Drugs and Medical Devices (BfArM) to seek Scientific Advice, in addition to, or in lieu of EMA Scientific Advice. Due to the significant time, effort and expense that may be spent on these procedures, it is crucial developers are able to obtain agency input at the right time in their development programme, and of the highest value possible.
What is Scientific Advice?
The scope of Scientific Advice encompasses all the key elements of a drug development programme, allowing the developer to ask specific questions directly to National Agencies or EMA concerning:
Quality (manufacturing, chemical, pharmaceutical and biological testing);
Non-clinical (toxicological and pharmacological tests);
Clinical aspects (studies in human subjects - either patients or healthy volunteers, including clinical pharmacological trials designed to determine the efficacy and safety of the product for pre- or post-authorisation activities including risk-management programmes);
Specific considerations regarding development for the paediatric or geriatric population (where applicable).
EMA Scientific Advice also allows discussion of issues relating to interpretation and implementation of draft and fully published EU guidelines. In the case of medicines targeted for Orphan indications, additional questions may be asked concerning the maintenance of Orphan status at the time of Marketing Authorisation Application (MAA) submission.
Scientific Advice also provides an opportunity to interact with the Paediatric Development Committee (PDCO) and pose questions in this regard, as an agreed Paediatric Investigation Plan (PIP) is a requirement in Europe ahead of submission of the Marketing Authorisation Application.
Strategy & Timing
In general, it is recommended that the developers of any medicinal product seek Scientific Advice at some point in the development programme. While Scientific Advice does not provide an avenue for pre-assessment of any data, it is a key tool for understanding the expectations of the regulatory authorities at different stages of development. Defining the questions to be asked, as well as the timing of the advice procedure, can greatly impact the outcome and the utility of the regulator’s advice. A clear regulatory strategy incorporating when advice is sought from the regulators is crucial to optimise a development programme. Deciding on seeking advice from the EMA or a national authority can also be driven by the intended route for eventual authorisation in the EU. For example, products which will be authorised via the EU’s centralised procedure would naturally benefit from advice provided by the EMA. However, even in this situation, National Competent Authority advice can still be of value, offering an opportunity to gain insight into the design and data requirements to support clinical trial applications.
National Scientific Advice Procedure
The procedure for seeking Scientific Advice from an NCA is less formal than that associated with seeking EMA Scientific Advice. The process by which an NCA should be approached to request Scientific Advice is well defined by each individual agency. Typically, a request for Scientific Advice should be submitted at least eight weeks in advance of the preferred meeting date. A finalised briefing pack and list of questions (along with a Company position statement to accompany each question) should be submitted ahead of the meeting in agreement with the respective agency. Unlike the EMA procedure, Scientific Advice from NCAs is often conducted as a face-to-face meeting. Following the meeting, the Applicant submits their minutes to the agency for review and confirmation.
The EMA Scientific Advice Procedure
EMA Scientific Advice is prepared by the Scientific Advice Working Party (SAWP) and formally issued by the Committee for Medicinal Products for Human Use (CHMP), the committee responsible for review of an MAA; in this sense, advice issued provides a good barometer of the expectations of the Agency for a positive opinion at the time of MAA. The EMA scientific advice procedure is commonly a fully written procedure associated with formal published submission timelines. In some cases, EMA may request a face-to-face discussion meeting with the applicant.
Defining an Appropriate Strategy
Although the variety of agencies from which to seek Scientific Advice may seem daunting, the EU regulatory landscape provides valuable opportunities to seek multiple agency opinions in advance of EMA interactions. Many applicants adopt a two-tier strategy ahead of Phase II, with particular focus on regulators for jurisdictions in which a Clinical Trial Application (CTA) will be filed. The exact timelines for seeking Scientific Advice will, of course, vary and are dependent on the nature of the individual development programme. For example, an applicant could choose to seek NCA advice prior to Phase II to garner input on the clinical study strategy and enter into EMA SA ahead of Phase III for advice on the MAA development strategy.
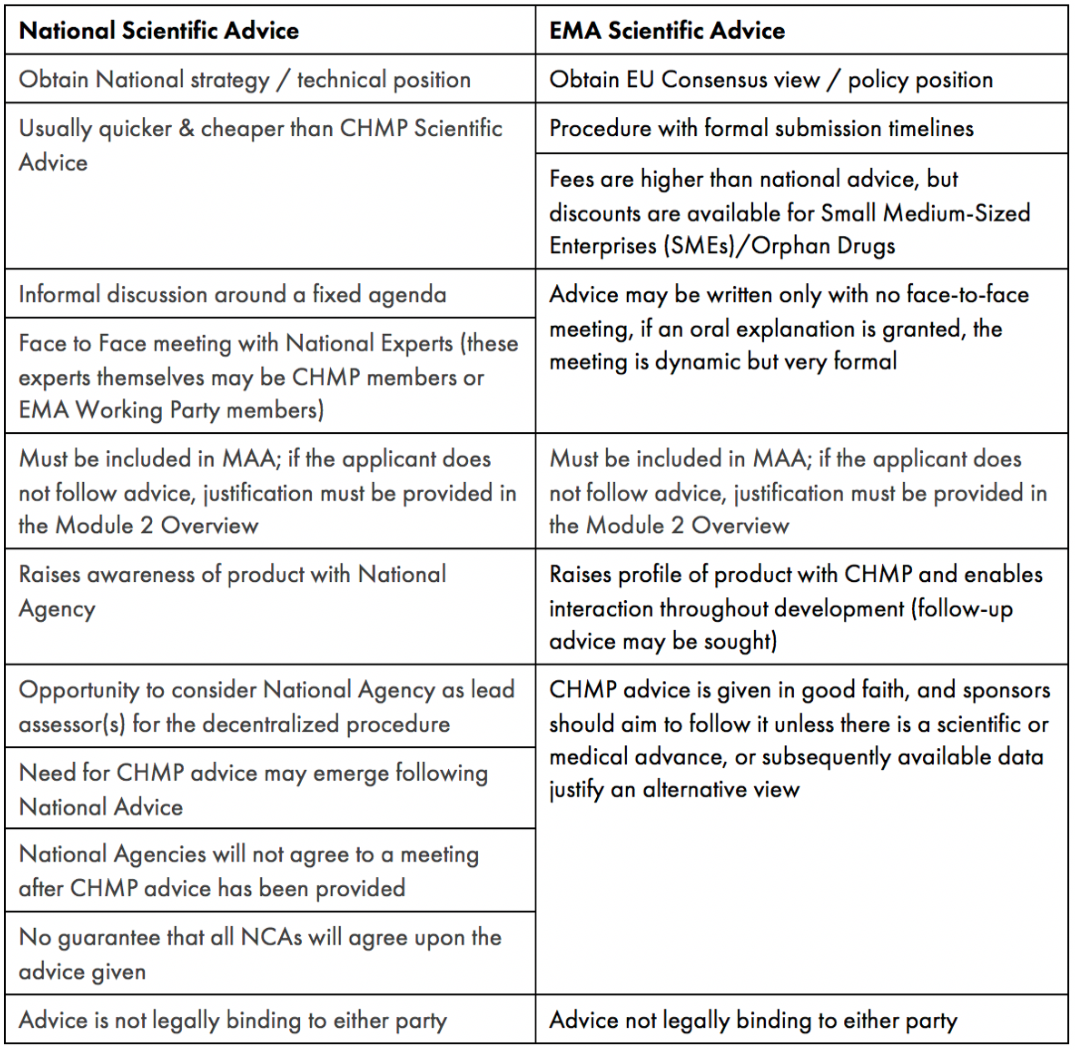
Table 1 provides a comparison on the main considerations of the Scientific Advice processes offered by NCA and EMA.
Table 1: Key Considerations for Seeking National and EMA Scientific Advice
Seeking Advice from a National Competent Authority (NCA)
In contrast to the rigid written advice procedure provided by EMA Scientific Advice, the face-to-face meeting format of most NCA advice meetings allows for a more dynamic and interactive discussion of guideline interpretation, scientific concepts and data. Crucially, being in the same room as the regulators provides an opportunity to be reactive to their comments and persuasive in the presentation of your data. In many cases a written procedure is also available, if the applicant prefers.
As EMA advice is provided solely in a written format, unless the EMA request a face-to-face discussion meeting, there is usually no opportunity to discuss the product and its development in person; a pre-submission meeting is available if desired, however, this is restricted to administrative details.
The timelines associated with NCA advice are less formal than those prescribed for EMA advice, with the applicant able to request meeting dates based upon their own availability. As CTAs are submitted for assessment at the NCA for the country in which the trial will be conducted, NCAs are well placed to provide advice on the specifics of the protocol, as well as the sufficiency of the data package to support trial approval.
While scientific advice garnered from national agencies does not always represent a pan-European view like EMA, it does provide a useful avenue for determining responses likely to be put forward during the EMA scientific advice procedure, allowing the applicant to pre-emptively address any items of particular concern, and optimise their briefing package for the more formal EMA written advice procedure. However, the Applicant should be aware that the number of questions that can be posed to any NCA is restricted by the length of the face-to-face meeting (typically 90 minutes), or, in the case of the French National Authority (ANSM) to 6 questions.
An additional point of consideration when obtaining advice from more than one NCA is that their responses to questions may differ from one another. For this reason, it is recommended that the applicant seek advice from three NCAs, using the same briefing package, to ensure a consensus can be reached on any contentious issues.
European regulatory agencies have recently taken steps to address this common issue through the implementation of the Simultaneous National Scientific Advice (SNSA) pilot programme. This procedure aims to provide consolidated advice from two national agencies on the basis of a single application and data package. Advice can be sought from any combination of participating NCAs. This pilot runs for one year from 1st February 2020, and the list of participating agencies is available here.

Why Choose a Certain National Competent Authority (NCA) Over Another?
An Applicant’s choice of NCA may be influenced by a number of factors. Chiefly, if the applicant is planning to hold a clinical trial in a certain country, the NCA of that country would be best placed to offer guidance on the approvability of that trial.
In order to obtain the highest possible quality of guidance, and be guaranteed a suitable level of responsiveness, it is recommended that Scientific Advice be sought from a large, well-funded agency such as those in Austria, Finland, France, Germany, Ireland, The Netherlands, Sweden (or the UK). The choice of agencies further narrows if an applicant wishes to make use of the aforementioned SNSA pilot programme, as not all NCAs are participating in this scheme.
Some companies may also wish to factor in the financial burden when choosing NCAs. The expense associated with Scientific advice varies greatly between agencies. For example, advice from ANSM is free while the Belgian Authority (FAMHP) charges almost €18,000 depending on the scope of the advice sought. In the scale of most drug development programmes, this fee may not seem prohibitive, but the key consideration here is value for money. For example, a company with SME designation would be expected to pay just €8,900 for full EMA Scientific Advice.
Typically, an NCA meeting can be scheduled within 2 to 3 months of submitting the meeting request; sometimes sooner, although this depends on the scope of advice (quality and/or nonclinical and/or clinical aspects), as well as the applicant’s flexibility in terms of meeting dates. These timelines vary based on the NCA chosen, particularly with regard submission of the briefing document, therefore it is recommended that the applicant familiarise themselves with the submission timelines as listed on the website of the relevant NCA.
Preparing for EMA Scientific Advice
In order to obtain the maximum value possible form agency interactions, the timing of an EMA Scientific Advice procedure is key. Although advice can be sought at any time, it is typically more common to wait until after Phase 1, when sufficient data are available to support development plans and formulate specific questions. Most frequently, advice is sought in relation to the Phase III development programme (Pivotal Studies) to support an eventual MAA. For products seeking an accelerated route to market, however, advice is frequently sought earlier or prior to Phase II.
As mentioned previously, EMA Scientific Advice is predominantly a written procedure with associated formal timelines. The standard duration for an advice procedure is 40 days, however, the SAWP could elect to request a meeting with the Applicant to seek clarification on any number of points prior to adoption of final advice. In such an instance, the so-called ‘70-day procedure’ would be followed. The subsequent discussion meeting must only address the points raised by the SAWP; the Applicant is not able to request a face-to-face meeting with the SAWP during the Scientific Advice process.
The formal EMA scientific Advice application procedure is outlined in Figure 1.
Figure 1: Flow Diagram of EMA Scientific Advice Procedure

EMA offers an optional presubmission meeting for developers that require feedback on the administrative aspects of their draft application.
While seeking a presubmission meeting may be unnecessary for some development programmes, it should be considered if the applicant wishes to discuss the following:
The content and scope of the questions posed.
The structure of the scientific advice request.
Identification of any additional issues that should be included in the scientific advice request.
Any regulatory questions that fall outside the scope of scientific advice.
Optimisation of the final application package based on a pre-submission meeting can ultimately improve the quality of the scientific advice given.
Fee Reductions
Agency fees for provision of scientific advice can be significant and run into the tens of thousands of Euros. As an incentive, the EMA offers financial, procedural and administrative assistance to companies defined as SMEs under the EC Recommendation (2003/361/EC, 2003) and EC Regulation (EC 2049/2005). Enterprises may be designated an SME if established within the European Community, and if they meet certain thresholds for headcount and financial criteria.
It is still possible for non-EU SMEs to obtain the benefits of EMA scientific advice fee reduction without the cost of setting up an EU entity. EU-based regulatory consultancies such as CNS can provide a service allowing clients to append to their own SME status. This simple, low-cost activity can be completed in 6 weeks or less and can confer a 90% reduction in Scientific Advice fees, a significant saving. In the event that SME status is obtained, scientific advice is followed, and a marketing authorisation application is not successful a conditional fee exception will apply.
Fee reductions are also available for products that have obtained Orphan Designation, as defined by EC 141/2000. While discussion of the requirements for obtaining orphan designation is outside the scope of this paper, once obtained, developers of orphan drugs are eligible for a 75% reduction in scientific advice fees, increasing to 100% if the developer also holds SME status at the time of the advice procedure.
Associated Procedures Relevant to Scientific Advice
EMA participates in the following additional advice procedures to assist developers of medicinal products:
Parallel EMA/US FDA Advice Procedure: Allowing applicants to request concurrent advice from EMA assessors and FDA reviewers concerning the development of their medicinal product. This procedure aims to increase dialogue between the two agencies in order to optimise the product development strategy. However, the advice of each agency may still differ.
Submission to Health Technology Assessments (HTA): Offering consultation in parallel with European Network for Health Technology Assessment (EUnetHTA). This process allows a developer to obtain feedback from regulators and HTA bodies on their evidence-generation plans to support decision-making on marketing authorisation and reimbursement of new medicines at the same time. The procedure is a single gateway for parallel consultations with EMA, EUnetHTA and HTA bodies on their evidence-generation plans.
Summary and Conclusions
Although appearing complicated at first, the availability of Scientific Advice in the EU from both EMA and individual NCAs provides the opportunity for medicinal product developers to obtain comprehensive, high quality guidance tailored to the specific needs of their development programme. Both EMA and NCA advice procedures have benefits and constraints, which must be considered alongside clinical development timelines in order to extract the most value possible from a given procedure.
While the appropriate Scientific Advice strategy may vary across development programmes, typically a two-tiered approach - first seeking advice from up to 3 NCAs in parallel - allows for optimisation of the briefing package questions and content before approaching EMA. Scientific Advice from EMA, if sought strategically, is critical to most drug development programmes, allowing an applicant to identify and address any issues ahead of pivotal trials and subsequently smoothing the road through MAA approval.