

US FDA Expedited Review Processes: Fast Track Designation and Breakthrough Therapy Designation.
by Iheoma Anosike
For a medicine to be approved, Sponsors need to demonstrate that it has a positive risk-benefit balance in the proposed target population, and is of satisfactory quality. These attributes must be shown throughout the product lifecycle and are rigorously assessed by regulatory agencies prior to approval of the medicinal product. Despite the requirement for drug development and review processes to be thorough and in-depth, it is also in the interests of public health that drugs are available to patients in a timely manner. This particularly holds true for patients suffering from serious and life-threatening diseases. Thus, it is important that regulatory agencies have procedures at their disposal, to incentivize and accelerate the development of medicines which target serious diseases, without negating patient safety and clinical benefit.
The US Food and Drug Administration (FDA) has four programs intended to facilitate and expedite the development of new drugs/biologics to address an unmet medical need in the treatment of a serious or life-threatening condition: Fast Track designation, Breakthrough Therapy designation, Accelerated Approval and Priority Review designation. A drug development program may qualify for more than one expedited program. It should be noted that these four drug development programs are not for advanced therapies; a separate program is available for such products. This paper is based on FDA’s 2014 Guidance: Expedited Programs for Serious Conditions – Drugs and Biologics and will specifically focus on Fast Track and Breakthrough Therapy designations, providing an overview of the procedures and requirements for these programs. Recommendations from Scendea’s team of expert regulatory consultants pertaining to the strategy and timing of requests for these designations will also be covered.
FAST TRACK DESIGNATION
Fast Track designation was introduced in 1997 as part of the FDA Modernization Act (FDAMA), and later amended in the FDA Safety and Innovation Act of 2012 (FDASIA). The program aims to facilitate the development and expedite the review of drugs and biologics to treat serious conditions and fill an unmet medical need.
Qualifying Criteria for Designation.
In order to be eligible for Fast Track designation, the proposed drug should be intended to treat a serious condition and nonclinical or clinical data must demonstrate the potential to address unmet medical need. Alternatively, the drug must have been designated as a qualified infectious disease product.
Determining whether a condition is serious is based on an assessment of whether the drug will impact factors such as survival, day-to-day functioning, or the likelihood that the condition, if left untreated, will progress from a less severe condition to a more serious one. To meet the requirement of filling an unmet medical need, the proposed therapy must provide an option for treatment where none exists or must be potentially better than available therapies.
To be successful with a request for Fast Track designation in a condition where there are already available therapies, the new treatment should fulfil at least one of following criteria:
Show superior effectiveness on serious outcomes or improved effect on serious outcomes.
Provide an alternative for patients not eligible or patients refractory to available treatments.
Show an improved safety profile compared to available treatments.
Can be used effectively with other critical agents that cannot be combined with available therapy and/or have a more favorable drug-drug interactions profile.
Improve compliance in a way that is expected to lead to an improvement on serious outcomes.
Improve the diagnosis of a serious condition where early diagnosis results in an improved outcome.
Address an emerging or anticipated public health need.
Sponsors should note that these criteria can be demonstrated using nonclinical or clinical data, depending on the product’s current stage of development. As well as nonclinical or clinical data, the mechanistic and theoretical rationale underlying the use of the product must be included in the request.
Features of Designation.
Sponsors awarded Fast Track designation for their medicinal product can expect to benefit from the following rewards associated with designation:
More frequent meetings with FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support approval. This includes pre-Investigational New Drug Application (IND), End of Phase (EOP) 1, EOP2, pre-New Drug Application (NDA) or pre-Biologics License Application (BLA) meetings.
More frequent written communication from FDA regarding topics such as the design of the proposed clinical trials.
Eligibility for Rolling Review if relevant criteria are met.
Eligibility for Accelerated Approval and Priority Review if relevant criteria are met.
Since the introduction of the Fast Track designation program, there has been an increase in the number of Fast Track products receiving approval, as shown in Table 1. Of the 187 drugs designated Fast Track by the Center for Drug Evaluation and Research (CDER) in 2020 alone, 36 obtained approval.
Table 1: Fast Track Designation Products Statistics Since Inception.

Adapted from (Chary, 2016)
Submitting a Request.
Fast Track designation requests can be submitted at any time during the drug development process, ideally no later than the pre-BLA or pre-NDA meeting. However, to receive the full benefits of the program, Scendea recommends that requests are submitted as early as possible in the drug development process (based on the availability of data required for the request), such as at the time of the initial IND submission. If the request is submitted after this time, Sponsors should note that requests must be filed as an IND amendment. Requests for Fast Track designation are submitted to Module 1, Section 1.7.1 “Fast track designation request” of the IND. As all submissions to an IND remain confidential, the FDA does not disclose Fast Track submissions or decisions, unless the submission has been publicly disclosed by the Sponsor.
FDA will review the request and decide within sixty days whether the drug meets the criteria for Fast Track designation. If the product is designated, a designation letter will be sent to the Sponsor outlining that Fast Track designation has been granted and that the development program must continue to meet the criteria for designation moving forward. If the Agency determines that a Fast Track designation request was incomplete or that the drug development program failed to meet the criteria for Fast Track designation, the Agency will send a non-designation letter to the Sponsor. The non-designation letter will state that fast track designation is not granted and explain the reasons for the Agency’s decision.
Contents of Designation Request.
Designation requests for Fast Track should include the following information. Scendea recommends that this information is captured as succinctly as possible, in approximately 10 to 20 pages.
If the Fast Track designation request is submitted to the Sponsor’s IND as an amendment, the submission must be identified in the cover letter as a “REQUEST FOR FAST TRACK DESIGNATION” in bold, uppercase letters.
If the request is submitted with an initial IND, the submission needs to be identified in the cover letter as both an “INITIAL INVESTIGATIONAL NEW DRUG SUBMISSION AND REQUEST FOR FAST TRACK DESIGNATION” in bold, uppercase letters.
In the cover letter of the submission, the name of the Sponsor’s contact person and the contact person’s address, email address, telephone number, and fax number.
If applicable, the IND application number.
If available, for drug products, the proprietary name and active ingredient and for biological products, the proper name and proprietary name.
The division or office to which the IND is being submitted or in which it is active.
The proposed indication(s).
A concise summary of information that supports the Fast Track designation request for the indication being studied, including the following:
o The basis for considering the drug to be one intended to treat a serious condition.o The basis for considering the drug to have the potential to address an unmet medical need and an
explanation of how this potential is being evaluated in the planned drug development program (e.g., a
description of the trials intended to evaluate this potential).If applicable, a list of documents previously submitted to the IND that is considered relevant to the designation request, with reference to submission dates.
BREAKTHROUGH THERAPY DESIGNATION
Breakthrough therapy was introduced under the FDA Safety and Innovation Act in 2012. The program is designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s).
Qualifying Criteria for Designation.
In order to be eligible for a Breakthrough Therapy designation, the drug should be intended to treat a serious condition and preliminary clinical evidence should indicate that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies. If there is no available therapy, the new drug should show a substantial and clinically meaningful effect on an important outcome over placebo or a well-documented historical control.
Assessments for the degree of improvement associated with the new drug over existing treatments is subjective but typically depends on both the magnitude of the treatment effect, which could include duration of the effect, and the importance of the observed clinical outcome. A clinically significant endpoint generally refers to an endpoint that measures an effect on irreversible morbidity or mortality (IMM) or on symptoms that represent serious consequences of the disease. A clinically significant endpoint can also refer to findings that suggest an effect on IMM or a serious symptom such as an effect on an established surrogate endpoint. In the Breakthrough Therapy designation request, a Sponsor should provide justification for why the endpoint or other findings should be considered clinically significant.
Features of Designation.
Breakthrough Therapy products are entitled to the features of the program listed below.
All Fast Track designation program features.
Intensive guidance on an efficient drug development program, beginning as early as Phase 1.
Organisational commitment involving senior
managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review.Eligibility for Rolling Review and Priority Review if relevant criteria are met.
Submitting a Request.
A Sponsor can receive preliminary Breakthrough Therapy designation advice from the review division to which an active IND is assigned, prior to the submission of a formal Breakthrough Therapy designation request.
To benefit from this, Sponsors must contact the regulatory project manager (RPM) in the relevant review division and request the “Preliminary Breakthrough Therapy Designation Request Advice” template. This template should then be submitted as a formal amendment to the IND and a subsequent teleconference between the sponsor and the review division will be set-up by the RPM. The review division will make a recommendation as to whether a request for a Breakthrough Therapy designation is appropriate, may be too preliminary, or does not currently meet the criteria for designation (FDA, 2021).
Ideally, a Breakthrough Therapy designation request should be received by FDA no later than the EOP2 meeting if any of the benefits of the designation are to be obtained. Sponsors are also encouraged to submit the Breakthrough Therapy request before initiation of pivotal clinical trials. Requests for Breakthrough Therapy designation should be submitted to Module 1, Section 1.12.4 “Request for Comments and Advice” of the IND. As all submissions to an IND remain confidential, the FDA does not disclose Breakthrough Therapy submissions or decisions unless the submission has been publicly disclosed by the applicant.
FDA will respond to Breakthrough Therapy designation requests within sixty days of receipt of the request. If the product is designated, a designation letter will be sent to the Sponsor outlining that Breakthrough Therapy designation has been granted and that the development program must continue to meet the criteria for designation moving forward. If the Agency determines that a Breakthrough Therapy designation request was incomplete or that the drug development program failed to meet the criteria for Breakthrough Therapy designation, the Agency will send a non-designation letter to the sponsor. The reasons for the Agency’s decision will be explained in the letter.
Cumulative data for the number of Breakthrough Therapy requests submitted to and granted by CDER and Center for Biologics Evaluation and Research (CBER) between 2012 – 2020, is shown in Table 2.
Table 2: Cumulative Data for Breakthrough Therapy Requests.

Adapted from (CBER, 2020; CDER, 2020)
When all products designated Breakthrough Therapies from 2012 – 2020 are considered, the number of cumulative CDER and CBER approvals for these products are 190 (51%) and 11 (22%), respectively.
Contents of Designation Request.
Designation requests for Breakthrough Therapy should include the following information. Scendea recommends that this information is captured in approximately 10 to 20 pages.
If the Breakthrough Therapy designation request is submitted to the Sponsor’s IND as an amendment, the submission should be identified in the cover letter as a “REQUEST FOR BREAKTHROUGH THERAPY DESIGNATION” in bold, uppercase letters. If the request is submitted with an initial IND, the submission should be identified in the cover letter as both an “INITIAL INVESTIGATIONAL NEW DRUG SUBMISSION AND REQUEST FOR BREAKTHROUGH THERAPY DESIGNATION” in bold, uppercase letters.
In the cover letter of the submission, the name of the Sponsor’s contact person and the contact person’s address, email address, telephone number, and fax number.
If applicable, the IND application number.
If available, for drug products, the proprietary name and active ingredient and for biological products, the proper name and proprietary name.
The division or office to which the IND is being submitted or in which it is active.
The proposed indication(s).
A concise summary of information that supports the Breakthrough Therapy designation request for the indication being studied, including the following:
o The basis for considering the drug to be one intended to treat a serious condition.
o The preliminary clinical evidence that the drug may demonstrate substantial improvement over available therapies.
FDA does not expect the submission of primary datasets, but the preliminary clinical evidence should
be described including a brief description of the study results and statistical analyses.If applicable, a list of documents previously submitted to the IND that is considered relevant to the designation request, with reference to submission dates.
Harmonized Development in EU and US.
Sponsors should note that whilst the European Medicines Agency (EMA)’s Priority Medicines (PRIME) scheme shares the same objectives as the FDA’s Breakthrough Therapy designation program, both designations have a different legal basis and as such, harmonisation between the two procedures is difficult.
It is possible for Sponsors to separately receive both Breakthrough Therapy designation and eligibility to PRIME (i.e. dual designation). EMA and FDA both encourage Sponsors to inform the relevant Agency to whom a request is submitted, whether they have submitted a request for designation or eligibility to the other Agency and the outcome of this request. In this manner, both Agencies aim to maintain an increased awareness of FDA/EMA dual designated products and stimulate early dialogue in both regions on an ad hoc basis, or in the context of the Parallel Scientific Advice (PSA) program via a formal meeting. For successful planning of global development programs, both Agencies encourage Sponsors to contact FDA and EMA on a dual designated product’s development program and seek joint advice under the PSA program. Sponsors wishing to nominate a product for a PSA procedure should address one single “Request for PSA” letter to both Agencies.
COMPARISON BETWEEN FAST TRACK
& BREAKTHROUGH THERAPY DESIGNATIONS
Table 3 provides a summary of the key features of both expedited development programs.
Table 3: Comparison of Fast Track and Breakthrough Therapy Designations for Serious Conditions.
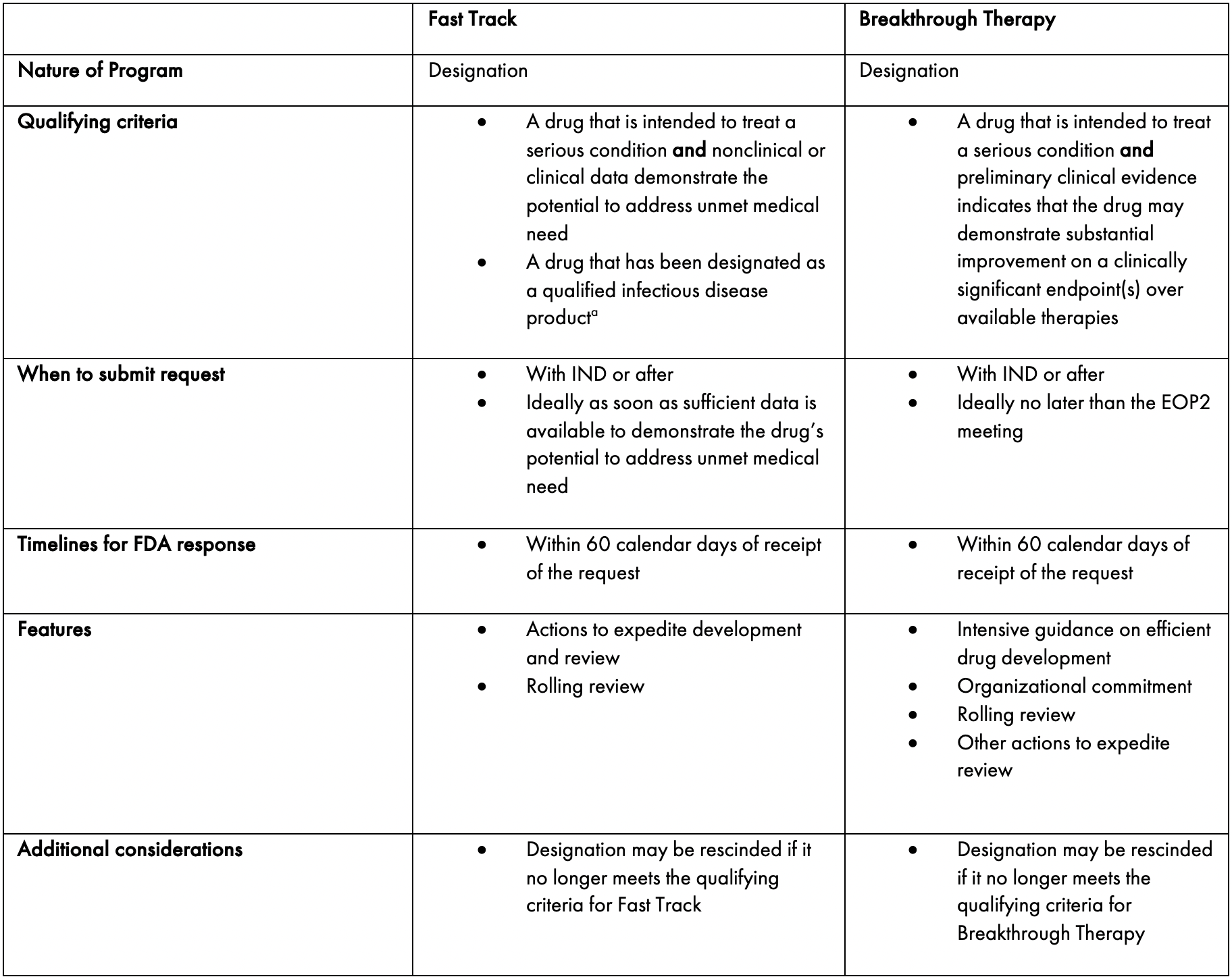
a. Title VIII of FDASIA, Generating Antibiotic Incentives Now (GAIN), provides incentives for the development of antibacterial and antifungal drugs for human use intended to treat serious and life-threatening infections. Under GAIN, a drug may be designated as a qualified infectious disease product (QIDP) if it meets the criteria outlined in the statute. A drug that receives QIDP designation is eligible under the statute for fast track designation and priority review.
Adapted from (FDA, 2014)
Even though both designations can be requested early in development, the requirements for Breakthrough Therapy designation are higher than those for the Fast Track program. Fast Track designation can be requested with nonclinical data and/or preliminary clinical evidence. However, for Breakthrough Therapy designation, clinical data must be available to demonstrate the benefit of the product over available therapies (or a placebo or historical control if there are no available therapies).
In addition, for Breakthrough Therapy designation, the improvement demonstrated must be substantial, while Fast Track designation requires only the potential for improvement.
Therefore, in deciding which of these designations to apply for, as well as considering the associated benefits, Sponsors must examine the requirements in light of the specific data package available for the product. Scendea often recommends that Sponsors initially apply for Fast Track designation and later submit a request for Breakthrough Therapy designation as development progresses.
Finally, products that qualify for Breakthrough Therapy designation receive more benefits than Fast Track products. Breakthrough Therapy designation provides the opportunity for earlier meetings and interactions on a more continuous basis throughout development in comparison to Fast Track designation. For example, Sponsors can access discipline-specific meetings outside of the critical IND milestone meetings for which the frequency can be determined between the Sponsor and FDA in a unique communication plan. In addition, Breakthrough Therapy products may receive greater access and coordination from FDA personnel (Kepplinger, 2015).
CONCLUSION
Both Fast Track and Breakthrough Therapy programs provide an opportunity for invaluable and tailored input from FDA throughout the product development program. This allows for a more efficient development program and eligible products are also more likely to access accelerated approval and priority review (if they qualify for these programs).
Scendea recommends that Sponsors select which of the designations would be most appropriate for the product and development program, considering the data available at the time of submitting the designation request. To obtain maximum gain from the benefits available for both programs, the request for Fast Track designation should be submitted as soon as robust preclinical (ideally pharmacodynamic) data is available for the product, whilst Breakthrough Therapy designation requests should be submitted once the required clinical data is available, ideally no later than the EOP2 meeting. Once a drug receives Fast Track or Breakthrough Therapy designation, early and frequent communication between the FDA and Sponsor is encouraged throughout the remaining drug development and review process. This will greatly increase the chance of earlier approval if the product qualifies for accelerated approval or priority review, and expedite patient access to the drug.
References.
CBER (2020). CBER Breakthrough Therapy Designation Requests Received by Fiscal Year.
CDER (2020). CDER Breakthrough Therapy Designation Requests Received by Fiscal Year.
Chary, K.V. (2016). Expedited drug review process: Fast, but flawed. J. Pharmacol. Pharmacother. 7, 57–61.
FDA (2014). Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics.
FDA, C. for D.E. and (2021). Frequently Asked Questions: Breakthrough Therapies. FDA.
Kepplinger, E.E. (2015). FDA’s Expedited Approval Mechanisms for New Drug Products. Biotechnol. Law Rep. 34, 15–37.