

Engaging With Regulators on Novel Statistical Approaches to Clinical Development.
in Collaboration with Exploristics.
Current Challenges in Drug Development
The Pharmaceutical Industry is undergoing significant change as it faces extreme pressure to deliver new treatments to market at a time of increased spending in R&D. Multiple economic pressures are being exerted on the industry, ranging from an impending patent cliff, a decrease in income due to greater competition from generics and biosimilars, to demands on drug pricing by governments. Each approved new drug costs around $3 billion to develop whilst healthcare providers seek to reduce the price paid for treatments making development costs unsustainable in the medium term. The current high attrition rate of treatments in development contributes to these high R&D costs, with 90% of treatments undergoing clinical trials failing to reach the market.
The current boom in life sciences has presented both an increase in opportunities as well as greater complexity, costs and uncertainty for large Pharma in delivering successful development programmes. The emergence of new clinical targets, cell therapies and methods of drug discovery has coincided with a reduction in the return on late-stage pipelines from 10.1% to 3.7% between 2010 and 2016 for the top 12 pharma companies as outlined by Deloitte. Moreover, the industry is experiencing a digital revolution in which life sciences are looking to harness technologies such as artificial intelligence (AI) and machine learning in addition to mining data from a wide range of real-world evidence sources, further adding to the complexity and investment now required for R&D programmes. Finally, the promise of precision medicine, personalising and targeting therapies to improve safety and efficacy has seen the demise of the blockbuster and forced companies to rethink pipelines and product development strategies.
Striving to maintain both profitability and agility in a rapidly changing market, pharmaceutical companies are increasingly outsourcing their R&D activities to third parties including academic institutions, biotech start-ups, and contract research organisations (CROs). Pharma has sought partnership with research area specialists offering niche expertise, services and tools to capture opportunities in emerging fields such as machine learning and precision medicine.
Consequently, the balance of innovation has swung towards smaller, more nimble players able to pivot to meet the demands of a rapidly changing life science sector. This is borne out by the transformation in the innovation landscape which has seen a 103% increase in the number of new molecular entities (NMEs) discovered by small biopharma View and has translated into 63% of novel drug approvals over the last 5 years.
Emerging Solutions Involving Data and Designs
Regulators are offering novel development options (accelerated approvals), and guidance around new trial designs. Both the US Food and Drug Administration (FDA)and the EU European Medicines Agency (EMA) have provided incentive programmes to developers in order to expedite the path to market. For example, in 2019 alone, 29 of the 48 novel drug approvals were designated in one or more of the expedited categories of Fast Track, Break Through, Priority Review and/or Accelerated Approval. In Europe, the EMA launched PRIME in March 2016 to provide developers with early and enhanced scientific support to medicines that have the potential to significantly address patients unmet medical needs. In its 2 year overview, 177 requests for PRIME eligibility were requested and 36 medicines accepted into the scheme. As of April 2020, 48 medicines are listed as accepted into the programme. Six medicines that had been granted PRIME status have successfully been authorised.
In the clinical development context, the use of new approaches and technologies has ultimately led to an explosion of data comprising many complex inter-relationships between risk factors, outcomes and treatment effects. However, so far, the collation of complex data has had modest success due to poor implementation and data handling skills exacerbated by unsuitable trial design and limited analysis tools. In many cases the designs of clinical trials do not account for the complexity of the data to be collected and often make simplistic assumptions about the sources of variability in patient responses.
In part, this is due to difficulty in accessing information that support the assumptions, but it is also due to the widespread use of established tools that encourage thinking in terms of making fixed assumptions that define the relationship between a single factor (e.g. treatment) and patient response. As the specification of these assumptions within a study protocol form the basis for defining study success, the current practice does not adequately facilitate the adoption of new approaches nor their application in reducing attrition in development.
Undoubtedly, there have been welcome developments in new trial design options that attempt to deal with the increased complexity: Stratified; Enriched; Adaptive; Seamless; Basket; Umbrella; Pragmatic trials are all important additions to the drug development toolkit. However, their utility and application will depend on the specific development scenario. With many options available, there is no one size fits all. Which one should you choose and how does the performance compare with alternative options?
On the data analysis side, there remains a challenge to identify the key results from complex data and to present those results in a way that supports interpretation and enables a thorough understanding of the data. Indeed, there are a range of analysis tools and strategies as well as visualisation techniques that can be used in this scenario. However, without adequately capturing or pre-specifying these approaches in a protocol and Statistical Analysis Plan (SAP) or ensuring the study is specifically designed for their use, then any results obtained may be considered exploratory or supportive at best.
The development of a protocol and clinical trial design is an exercise in maximising the probability of making the right decision: a study that fails when there is no relevant treatment effect or a study that succeeds when there is. This is a multi-factorial optimisation process comprising the selection of the right study population characteristics, sample size, sampling schedule, stratification, endpoint(s) and observation time, analysis strategy and decision criteria. Many of the technological solutions help to address specific elements of this process and may be able to make incremental improvements to the likely success of the study.
However, there is a need to provide information in the right context to help decide which approaches or combinations are likely to be the most effective. Getting the right combination of factors has a multiplicative effect, making a massive difference in the success of the study.
The Increasing Importance of Statistics in the New R&D Paradigm
The new drug development paradigm highlights the importance of biostatistics. Statisticians have been an integral part of drug development for some time but now their role is pivotal to the success of a development programme. Over many years they have developed the scientific rigour to understand how to mitigate risks due to bias, uncertainty and sources of variability through design and analysis. Their logical, objective approach help to bring clarity to protocols and statistical analysis plans. Now, more than ever, statisticians need to be collaborative and consultative, getting involved at the earliest stages in protocol development to synthesise information from multiple domain experts and data sources. Their understanding of the regulatory landscape allows them to identify the most expeditious way to develop a drug through exploratory, translational and regulatory research with smarter design strategies. They are uniquely placed to implement objective, quantitative methods to evaluate the performance of new approaches and identify the best development strategy. Today’s statistician provides a vital function in terms of helping to guide strategic development decisions. Early engagement is clearly important but equally, statisticians also need access to information and a toolbox of modelling and simulation capabilities to rapidly evaluate the performance of design and analysis scenarios.
The implementation of modelling and simulation takes time but the return on investment is huge. Based on case studies we have supported over a number of years, early engagement and the use of a flexible modelling and simulation platform has delivered impressive results in terms of reducing the risks, costs and duration of clinical development.
Examples of the benefits have included:
Increasing the probability of success for a development programme by almost three-fold without adding to cost.
Increasing the probability of success for a precision medicine study by 41% without increasing the cost.
Reducing the development time by 4 years.
Saving US$20M on a single study.
Terminating an extensive development programme that had little chance of success.
Moreover, modelling and simulation offer further benefits such as supporting and facilitating collaboration: all stakeholders have an input and are able to agree on the evidence-generation package and the decision-making framework. This allows options under consideration to be ruled in or out very quickly, supporting the rapid, informed development of protocols. In turn, protocols are clear and unambiguous, leading to more definitive outcomes.

Engaging with Regulators
Adopting novel approaches and clinical protocols in drug development, although able to pay dividends, does not come without inherent risk. Regulatory authorities frequently advocate adoption of such approaches to increase the development of drugs, particularly for drugs intended to meet serious and life threatening conditions. In this context, the authorities are willing to engage with companies during the course of development to ensure appropriate data is generated to allow approval. As mentioned above, both FDA and EMA have introduced specific programmes to engage with companies in order to accelerate drug development.
To benefit from these programmes, understanding the mechanisms for engaging with the regulatory authorities is crucial. Through these interactions, companies are able to get agreement and advice from the regulators in order to implement novel development strategies.
In the US, the FDA under PDUFA legislation are required to offer formal meetings requested by companies who seek advice relating to the development and review of investigational new drugs and biologics, and drug or biological product marketing applications. Since these meetings represent critical points in the regulatory process, formal guidance to ensure efficient and consistent use of resources is available. The FDA offers three meeting types:
Type A: those that are necessary for an otherwise stalled product development programme to proceed or to address an important safety issue.
Type B: meetings undertaken at specific points such as PreIND, pre NDA/BLA. It is at these meetings where novel strategies for development, including provision of data for review should be discussed. Notably, Type B meetings are held to discuss the overall development programme for products granted breakthrough therapy designation status, Fast Track and Accelerated Approval.
Type C: any other meeting which does not fall under Type A/B.
The FDA meetings and advice may be provided either face to face with Agency reviewers, or more commonly for Type B meetings via telecons or written advice. For all three meeting types, strict timelines as outlined in relevant guidance are adhered to, and the company should provide comprehensive information by way of briefing documentation for review and comment. Clearly this documentation must be well prepared, concise and present relevant data to justify the developers clinical development strategy.
In Europe, to facilitate dialogue, the EMA provides a formal process for interaction through Scientific Advice under Article 57-1 (n) of Regulation (EC) No 726/2004. Unlike the US, developers also have another process to obtain advice from the National Competent Authorities (NCA) . such as the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA), or Germany’s Federal Institute for Drugs and Medical Devices (BfArM). Deciding on the appropriate route for meeting to discuss novel clinical methodologies with European regulators (EMA vs NCA) should be taken based on the ultimate route for authorisation of the product. Those medicines which will use the EU’s centralised procedure are strongly advised to discuss these topics directly with the EMA, where pan EU agreement on the strategy can be reached. If a national/decentralised route to market is envisaged, then NCA advice on this subject is likely to be more relevant. While Scientific Advice does not provide an avenue for pre-assessment of any data, it is a key tool for understanding the expectations of the regulatory authorities. The key attributes of NCA vs EMA Scientific Advice are outlined in Table 1. It should be noted, that if NCA advice is sought, companies are now offered a pilot programme for Simultaneous National Scientific Advice (SNSA).
EMA Scientific Advice is prepared by the Scientific Advice Working Party (SAWP) and formally issued by the Committee for Medicinal Products for Human Use (CHMP), the committee responsible for review of an MAA. Access to early scientific advice with the EMA is one of the key advantages to PRIME eligibility as it provides the opportunity for a company to agree complex strategies with regulators sooner than would normally be anticipated.
Table 1: Key Considerations for Seeking National and EMA Scientific Advice
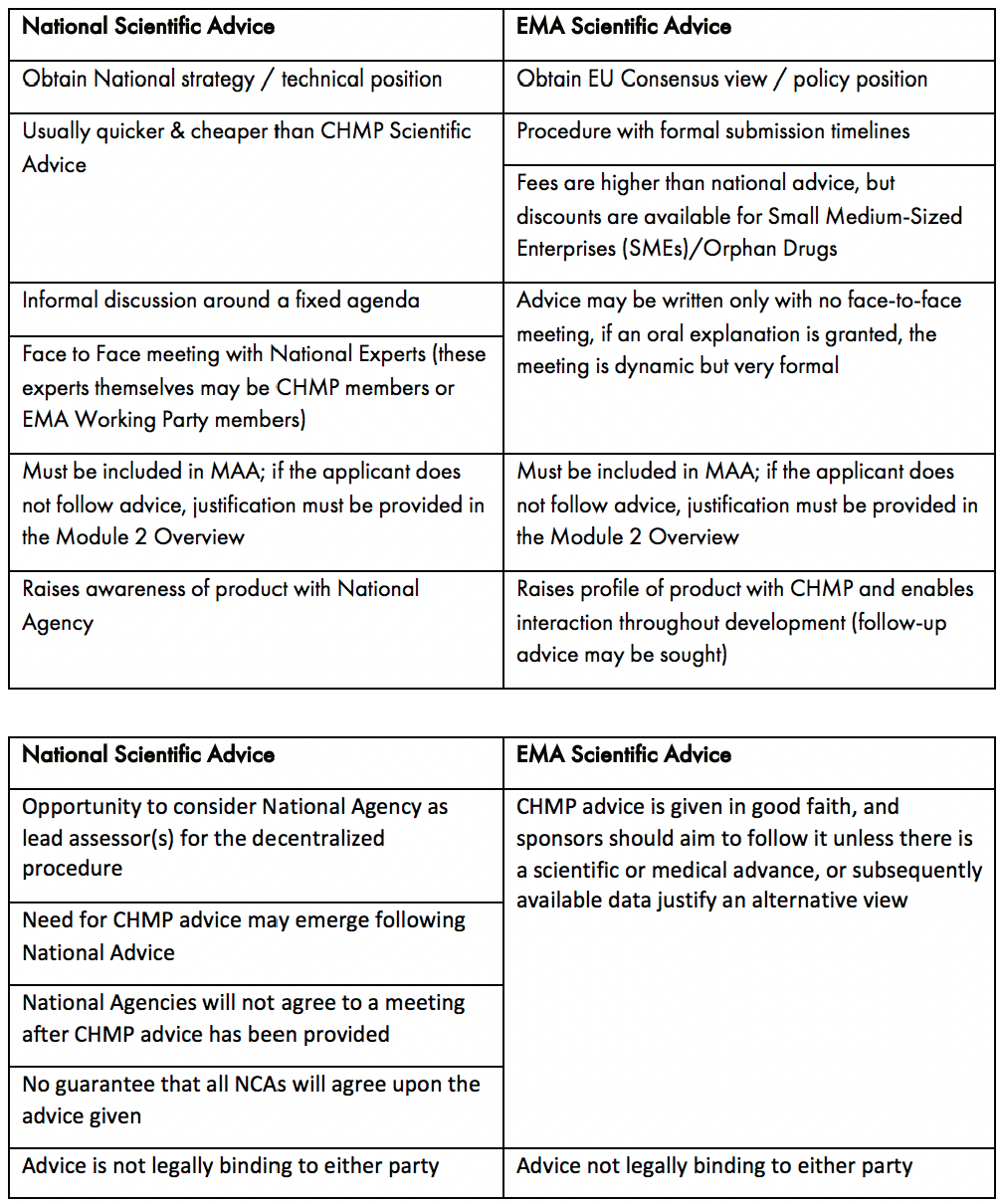
As with the US process, guidance on processes and required documentation is available from the regulatory authority. For the EMA, the process is driven according to specified submission deadlines published on their website. For NCAs, requests are submitted and dates for meetings are agreed between the developer and the respective NCA.
Conclusion
Overall, when using novel approaches to clinical development, seeking advice from the regulatory authorities, be it FDA, EMA or both, increases the likelihood of success. Ultimately, of course, it will be the quality and strength of the data generated by the clinical study which will determine the approvability of a drug candidate.