

Clinical Trial Regulation:
A New Chapter for EU Clinical Trials
In Collaboration with FGK.

Authors:
FGK
Dr Uwe Kramer
Scendea
Amy Cooke & Dr Maria Beatrice Panico
What is The Clinical Trial Regulation?
The “Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC”, in short, the Clinical Trials Regulation (CTR), regulates interventional clinical trials in EU Member States and European Economic Area (EEA) countries.
The CTR replaces Directive 2001/20/EC, the first EU-wide legislation for clinical trials of medicinal products which applied from 01 May 2004. In the EU, legal texts called “directives” are not immediately applicable. Directives define a framework of rules that come into force only after implementation into national law. This provides space for national interpretations and can ultimately result in a lack of harmonisation. By the end of the 2010s, there was considerable discontent with the lack of EU harmonisation and with the fact that approval had to be obtained in each of the participating EU Member States and not at EU level. When amending the CT legislation, EU legislators decided to opt for a regulation. Regulations apply immediately to all EU countries as soon as they enter into force and do not need to be transposed into national law, except for issues where the text of the regulation either explicitly delegates to the EU Member States, or with respect to an area that the regulation is silent about.
For example, Article 76 of the CTR demands that “EU Member States shall ensure that systems for compensation for any damage suffered by a subject resulting from participation in a clinical trial conducted on their territory are in place in the form of insurance, a guarantee, or a similar arrangement”. However, the CTR does not elaborate on this. The insurance cover may vary between EU Member States. Some EU Member States do not define a minimum insurance cover at all, and some may suggest that no insurance is required if it is replaced by some other type of guarantee.
The EU commission started a public consultation on the revision of the Directive 2001/20/EC in early 2011, issued a draft text for the CTR in July 2012, and in May 2014 the final text of the CTR was published in the “Official Journal of the European Union” and came into force the following month.
However, the CTR did not become applicable until 2022 due to delays implementing Article 99 of the CTR, which concerned the functionality of the EU portal and the EU database for clinical trials, now called the “Clinical Trials Information System” (CTIS). On the 31 July 2021 the EU Commission confirmed that CTIS was fully functional. The CTR came into effect six months later, that is from the 31 January 2022 onwards as mandated by Article 99. As a result of these delays, the CTR has essentially been implemented almost a decade after being authored.
After the first year of implementation all new trials will have to be conducted under the CTR. This means that from the 31 January 2023 sponsors will need to use CTIS to apply to start a new clinical trial in the EU/EEA. Sponsors will have until the 31 January 2025 to complete the transition of ongoing trials from the CT Directive to the CTR.
“Medicinal Product” is the EU legal term for products which may also be called medicine, drug or pharmaceutical elsewhere.
In the EU, “Medicinal Product” includes small molecules, biologicals, cell and tissue therapeutics as well as gene therapy products.
“Clinical Trials” refers to interventional studies of medicinal products. The CTR definition for “clinical trials” in Article 2 of the CTR is similar to that in section 1.12 of ICH E6 (R2), but the CTR explains what makes an ICH-defined clinical trial an interventional trial.
What is Regulated by The CTR?
Only interventional trials fall under the scope of the CTR because these are the only type of trials requiring prior review and approval from regulatory authorities.
The CTR contains clear definitions of what constitutes a clinical trial falling under the scope of the CTR. Non-interventional studies are not within the scope of the CTR.
There is a thin, and sometimes blurred line, between a clinical trial and a non-interventional trial. For example: If the assignment of a trial participant to a particular medicinal treatment is decided before enrolment into the trial, and does not fall within normal clinical practice of the EU Member State where the trial is conducted, then the study is a clinical trial (if some other conditions are also fulfilled). From a regulatory perspective a study where the trial participants received a medicinal product in a previous trial and are followed-up to assess long-term safety and efficacy of that drug is still an interventional trial. This trial would fall under the scope of the CTR even if no medicinal product were to be administered during the course of the trial itself. Correct classification of a trial has legal implications: conducting an interventional trial without prior authorisation from the competent authorities is against the law. It is recommended that Sponsors assess each trial carefully and take into account the local health system; if a Sponsor is in doubt whether the trial is interventional or not, they should seek advice in order to avoid lack of legislative compliance.
The CTR includes provisions for the approval of clinical trials and substantial modifications, for notification requirements, publication of trial documents and data and for trials in vulnerable subjects, et cetera. It also lays out detailed requirements for the trial protocol, the labelling and lay summaries of the results. It describes relevant aspects of the patient information sheet, the consent form, the investigator’s brochure and the annual safety report, et cetera.
Documentation That Can Be Used to Support
a Clinical Trial Authorisation Application.
The CTR clarifies which documentation can be used to support a clinical trial authorisation (CTA) application. For example, the CTR states in Article 25 that the non-clinical information presented in the application dossier must derive from studies complying with EU principles of good laboratory practice. In addition, clinical data generated in trials conducted outside the EU can be used to support the conduct of a trial in the EU only if the non-EU trials have been conducted in accordance with good clinical practice principles as defined in the CTR. Moreover, data from a clinical trial starting after 30 January 2022 is only accepted if that clinical trial has been registered prior to its start in a public register that is a primary or partner registry of, or a data provider to, the WHO International Clinical Trials Registry Platform (ICTRP).
The Sponsor: One, More & an EU Representative.
Unlike the Directive 2001/20/EC the CTR allows the presence of multiple trial sponsors (see Articles 71 and 72). In case of co-sponsorship, all co-sponsors “shall have the responsibilities of a Sponsor set out in” the CTR, “unless the sponsors decide otherwise in a written contract setting out their respective responsibilities”. However all co-sponsors need to agree on some points such as: assigning one of the sponsors for the application procedures, assigning one of the sponsors for being a contact point and assigning one of the sponsors for implementing the corrective measures that are ordered by the EU Member States.
The Directive 2001/20/EC established the role of the EU legal representative of a sponsor not established in the Union, but was somewhat silent about its responsibility. The CTR states that “such legal representative shall be responsible for ensuring compliance with the sponsor’s obligations pursuant to [the CTR] and shall be the addressee for all communications with the sponsor provided for in [the CTR]”.
The Purpose of CTIS and What it Replaces.
As described in Article 81, CTIS is the online platform that enables interactions between Sponsors and EU Member States concerning authorisation of clinical trial and substantial modifications, notifications and publication of trial-related documentation. However, not all regulatory activities of a clinical trial involve CTIS: SUSAR reports are still sent to the EudraVigilance database (which is linked to CTIS). Some aspects of clinical trials that are country-specific are not to be documented in CTIS at all (e.g., the authorisation of trial-specific radiological examinations in Germany or the notifications to the French data protection agency).
CTIS replaces both EudraCT, the EU’s non-public database for clinical trials and the public EU Clinical Trials Register. EudraCT and the EU Clinical Trials Register will remain operational to support trials conducted under the Directive 2001/20/EC. It has also been announced that the EU Clinical Trials Register will remain available as a legacy database.
Overview of CTIS.
MANAGEMENT: ORGANISATION-CENTRIC VS. TRIAL-CENTRIC
There are two management options for trial within CTIS: organisation-centric and trial-centric. The organisation-centric approach is recommended by EMA for Sponsors that run multiple clinical trials. It operates in a hierarchical manner whereby there is an overall Sponsor administrator who creates CTA applications and then assigns a specific clinical trial administrator to each trial, who in turn assigns users who will be working on the specific trial. Users working on a trial may be within the Sponsor organisation or external users from a contract research organization (CRO) or consultancy (see below). The second option, trial-centric, allows any user to create a clinical trial and become an administrator and assign other users to work on that clinical trial. With this second approach, there is no overall oversight and no control by an organisation on the trials that its employees create. It is expected that the majority of Sponsors will use the organisation-centric approach and only small organisations or academic sponsors will avail themselves of the trial-centric approach.
WAYS FOR SPONSORS TO WORK WITH CONSULTANCIES/CROS
Sponsors do not have to submit their CTA applications by themselves; they can delegate this task to consultants or CROs. To be able to submit CTA applications, sponsors and subcontracted applicants have to register themselves with the online portal CTIS. A prerequisite for registration with CTIS is the registration on EMA’s “Organisation Management Service”. Persons who want to be involved in regulatory procedures with CTIS must be given one or more of the pre-defined roles in CTIS. Concerning these roles, the online portal CTIS offers great flexibility. CTIS offers up to 18 roles for entities associated with the sponsor, e.g., CROs and clinical trial centres. These roles are not defined in the CTR, but in the comprehensive training material that is provided on the EMA website. To understand the concept of these roles, it is recommended to read the “FAQs - Management of roles and permissions”, a part of Module 7 of the CTIS training program.
Documents to Support a Clinical Trial Authorisation (CTA) Application.
Under the CTR the assessment of a CTA application will be divided into Part I and Part II.
According to Annex I of the CTR, Part I of the application dossier contains the following documents (as applicable): cover letter, EU application form, protocol, Investigator’s Brochure (IB), Good Manufacturing Practice (GMP) compliance documentation, Investigational Medicinal Product Dossier (IMPD) or simplified IMPD, auxiliary medicinal product (AxMP) dossier, Scientific Advice letter, Paediatric Investigation Plan and labelling. The AxMP dossier is a new requirement set in the CTR. According to Article 2 of the CTR, an AxMP is defined as “a medicinal product used for the needs of a clinical trial as described in the protocol, but not as an investigational medicinal product”. It corresponds to what was previously called non-IMP. AxMPs are usually licensed drugs; in this case the AxMP dossier will be the Summary of Product Characteristics. If the AxMP is not authorised the AxMP will follow the same requirements as for an IMPD. The remaining documents required for Part I are not new elements, but Sponsors should be aware of new requirements for these documents.
An example is labelling, where there are some new requirements included in Annex VI of the CTR. The document requirements are the same for all EU Member States. However, some documents must be translated into local languages. They are the cover letter, protocol synopsis, patient information sheet, informed consent form and the labelling of the IMP(s).
According to Annex I of the CTR, Part II of the application dossier contains patient-facing documents, documents concerning the clinical trial sites documentation on insurance cover or other systems for indemnification and a statement by the Sponsor on compliance with EU data protection rules. Depending on the EU Member State elements of Part II of the application dossier may be new. For some EU Member States documentation about the suitability of the clinical trial sites and a statement on compliance with EU data protection rules are new requirements.
On the one hand the list of documents applies to all EU Member States. On the other hand the EU-wide harmonisation ends at this point. While the EU Commission has published templates for some of the items of Part II, it is clear that some of them will not be accepted in all EU Member States. For example, the EU Commission has published a two-page “Site Suitability Template”, which is supposed to be filled for each clinical trial site. The Spanish competent authority AEMPS presents on their website a one-page statement for the same purpose. On the other hand, in the Netherlands, the “Site Suitability Declaration” consists of a 16-page booklet. While in Germany the reviewers want to see at least a draft of the agreements between the Sponsor and the clinical trial sites, in Spain the Sponsors simply have to add a budget table called “memoria económica”.
The patient information sheet and the consent form cannot be harmonised. Even under the rules of the CTR, it is not possible to prepare one master text and have it simply translated into the languages of the EU Member States that participate in the clinical trial. Some EU Member States might only accept their standard structure and standard wording. In Germany, the working group of medical ethics committee has published a master text that fills 19 pages – and information about the clinical trial has yet to be added. The empty master text for the Netherlands has 29 pages and features a considerably different structure.
In Spain, such booklets would not be acceptable: the Spanish master text is 12-page long and the final text must not exceed 15 pages, however the Sponsor must then also prepare five pages on data protection.
In addition to the Part I and Part II documents listed above, the submission package must include the following information: proof of payment, deferral of publication dates of documents, information on the EU Member States concerned and proposed reporting Member State. The reporting Member State is responsible for coordinating the assessment of multinational clinical trial applications.
The deferral of publication dates of documents is an important consideration for Sponsors. Under the CTR, the default option when filing the CTA application will be for all information to be made public with the following exceptions:
Quality information, including the quality sections of the IMPD and any quality-related requests for information (RFIs);
Draft assessment reports;
Personal data of participants of the clinical trial; and
Financial agreements...
However, Sponsors have the option to request a deferral of publication for certain categories of information, as summarised in the table below.
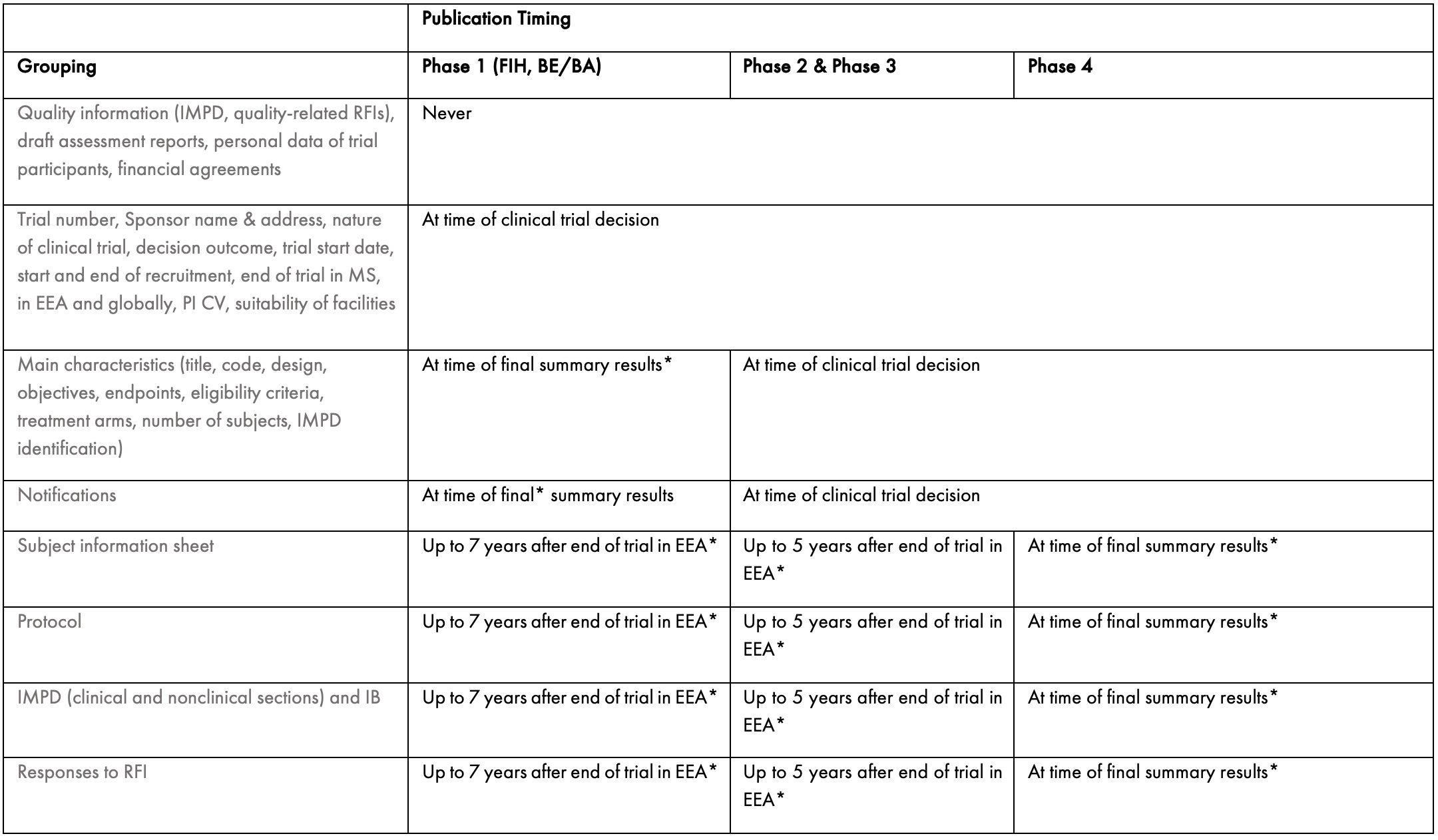
BA: bioavailability, BE: bioequivalence, EEA: European Economic Area, FIH: first in human, IB: Investigator Brochure, IMPD: investigational medicinal product dossier, MS: Member State, PI: Principal Investigator, RFI: request for information
* This timing is applicable if a request for deferral is accepted.
Redaction of The Documents.
Most documents that are submitted to CTIS will be published and therefore two version must be uploaded at the time of the CTA application: the original version and a redacted version for publishing. Guidance is available about which documents will be published and what information should be edited. Sponsors should remove commercially sensitive information; they also must remove any personal data leading to identification of trial participants as well as Sponsor’s representatives (i.e. names, birth dates, addresses, etc) in order to comply with the EU General Data Protection Regulation.
It should also be noted that, as joint controllers in terms of the EU General Data Protection Regulation, Sponsors will be responsible for any unintended disclosure of personal data through errors in redacting documents. Redaction and control of redaction demand a considerable amount of expertise and work and needs to be planned before submitting any CTA application via CTIS.
CTA Application: Process & Timelines.
Sponsors may choose to submit Part I and Part II either sequentially or in parallel. When the sequential approach is taken, Part II must be submitted within 2 years of approval of Part I. For a trial in multiple EU Member States a mixed approach can be taken, whereby Part I is submitted to all EU Member States participating in the clinical trial and Part II only submitted to some.
The CTR specifies timelines applicable to regulatory authorities and those pertaining to Sponsors. Please refer to Figure 1 for an overview of the timelines.
It should be noted that the regulators assessment time may be extended by up to 50 days for clinical trials concerning Advanced Therapy Medicinal Products, biologics, medicines falling under the mandatory scope of the centralised procedure and orphan medicinal products. However, Sponsors will always have only 12 calendar days to reply to requests for further information (RFIs).
The CTR also clarifies the timelines for completing the validation of submitted applications. If the regulatory authorities have comments or believe that the application is not complete, Sponsors will have up to 10 days to comment and/or provide additional information.
According to the CTR, the approval process should follow a relatively predictable schedule of events. At the moment this is not the case. The “Key performance indicators (KPIs) to monitor the European clinical trials environment” show that from January 2022 to July 2022 decision times varied between 5 and 117 days. In addition, as of the end of July 2022, more than 50% of clinical trial applications did not make it to an authorisation. Few were actually rejected; in most cases the Sponsors withdrew applications or they failed to provide a response to the RFIs on time and the application was deemed to have lapsed.
Upon rejection, the sponsor has three options: to initiate an appeal procedure in the EU Member State concerned, to modify the application dossier and resubmit, or to avoid conducting the clinical trial in the Member States that rejected the application.
The use of the appeal procedure is limited to decisions of individual EU Member States and is not applicable to decisions of the reporting EU Member State.
Since many deficiency letters are published via CTIS (apart from those on quality issues in order to avoid disclosure of commercially sensitive information), sponsors may be able to learn about typical findings and improve the quality of their trial documents in advance of submission.
Figure 1.

Addition of a New EU Member State.
According to Article 14 of the CTR, an application to extend the clinical trial to an additional EU Member State may be submitted following the initial authorisation. The new Member State has 52 days for the initial assessment period. If the additional EU Member State requests additional information on Part I or Part II, the Sponsor must provide it within 12 days and the additional EU Member State has 19 days for an additional assessment. These 12 plus 19 days are added to the 52 days of the initial assessment of the application. It should be noted that the new Member State may only raise RFIs on Part I on the following grounds:
When it considers that participation in the clinical trial would lead to a subject receiving an inferior treatment than in normal clinical practice in the Member State concerned;
Infringement of its national law as referred to in Article 90;
Considerations as regards subject safety and data reliability and robustness submitted under paragraph 5 or 6.
In this scenario, the new members state will review the RFI response together with all other Member States concerned.
If the Sponsor does not provide the additional information within the 12 days or if the EU Member State deems the additional information to not be compliant to its request, or the ethics committee in that additional EU Member State issues a negative opinion, then the additional EU Member State may refuse the authorisation of the clinical trial on its territory.
Clinical Trial Maintenance.
SUBSTANTIAL MODIFICATIONS
The CTR has detailed provisions for obtaining authorisation of substantial modifications of Part I or Part II of the application dossier.
If an application for a substantial modification of the application dossier is complete, and if the reporting EU Member State (for Part I modifications) or the EU Member State concerned (for Part II modifications) does not ask for additional information, then a Part I substantial modification has to be authorised within up to 49 days [according to Articles 18 (3), 19 (1) and 20 (5) of the CTR]. A Part II substantial modification has to be authorised within up to 44 days [according to Articles 17 (2) and 20 (5) of the CTR]. However, these timeframes are extended by up to 15 days if at the validation stage the application is not considered complete [according to Article 17 (4) of the CTR] and by up to 31 days [according to Article 18 (6) and 20 (6) of the CTR] if the sponsor is asked to provide additional information during the assessment phase. If timelines are extended the Sponsor has up to 10 days to complete the application during the validation phase and up to 12 days to provide additional information during the assessment phase. As such, the maximum time period for the authorisation of a substantial modification is up to 95 days for a Part I modification and up to 90 days for a Part II one. It should be noted that for clinical trials conducted under the Directive 2001/20/EC, the review of an amendment application did not generally cause validation issues or requests for additional information. Therefore, it is expected that substantial modification procedures should be mostly completed within the 49 day (Part I) and 44 day (Part II) timeframes.
One of the main changes introduced by the CTR compared with the Directive 2001/20/EC is that parallel submission of substantial modification is not permitted under the CTR. If a substantial modification is under evaluation, another substantial modification impacting the same part of the trial (Part I, Part II, or both) cannot be submitted. Sponsors are encouraged to plan carefully when and what type of modification requests they submit. The CTR includes no provisions for cases in which the second amendment is urgent. However, a potential way around this is to withdraw the first modification to enable submission of a more urgent amendment.
NOTIFICATIONS
Under the CTR sponsors need to submit notifications that were not required when conducting trials under the Directive 2001/20/EC. The notifications summarised below must be submitted to the participating EU Member States via CTIS within 15 days of the event occurring in each EU Member State.
Start of clinical trial
First subject enrolled
End of recruitment
It is important to note that a clinical trial authorisation ‘expires’ if the trial does not start within 2 years of the authorisation itself. If this happens, the sponsor must submit a new CTA application.
ANNUAL REPORTS
According to Article 43 of the CTR, the sponsor must submit a report on the safety of each IMP used in the clinical trial annually. The annual report is the terminology used in the CTR to refer to the Development Safety Update Report (DSUR). In the EU some requirements for the annual report go beyond than what is described in the ICH Topic E2F guideline. They are explained in the Questions & Answers document that accompanies the CTR . The provision in Article 43 of the CTR states that the reporting obligation for annual reports ends with the final clinical trial conducted by the sponsor with the IMP.
End of Trial.
NOTIFICATIONS, E.G. NEED TO NOTIFY WHEN TRIAL ENDS IN MEMBER STATE, IN EU AND GLOBALLY
Article 2 of the CTR defines the “end of a clinical trial” as “the last visit of the last subject or at a later point in time as defined in the protocol”. Article 37 of the CTR provides clarification of which notifications are needed. Not only does the global end of the trial have to be notified, but also the end of the trial in each of the EU Member States participating in the clinical trial and the end of the trial in all third countries in which the clinical trial has been conducted. For each of these notifications the Sponsor has 15 days from the time the event occurred to communication via CTIS.
To avoid non-compliance with the notification requirements, the trial protocol should include a definition of what constitutes the end of the clinical trial in each EU Member States, in all EU Member States and in all participating countries, if there are non-EU countries.
RESULTS SUBMISSION
Sponsors are required to submit a summary of the results from studies within one year of the trial ending in all EU Member States. If this isn’t possible, for example the trial is still ongoing in non-EU countries, the summary results should be reported as soon as possible. In addition, Sponsors must provide a summary that can be understood by a layperson. The content of this summary is detailed in Annex V of the CTR and in a comprehensive guideline issued by the EU commission. The timing for publishing the summary results is outlined in the table below.
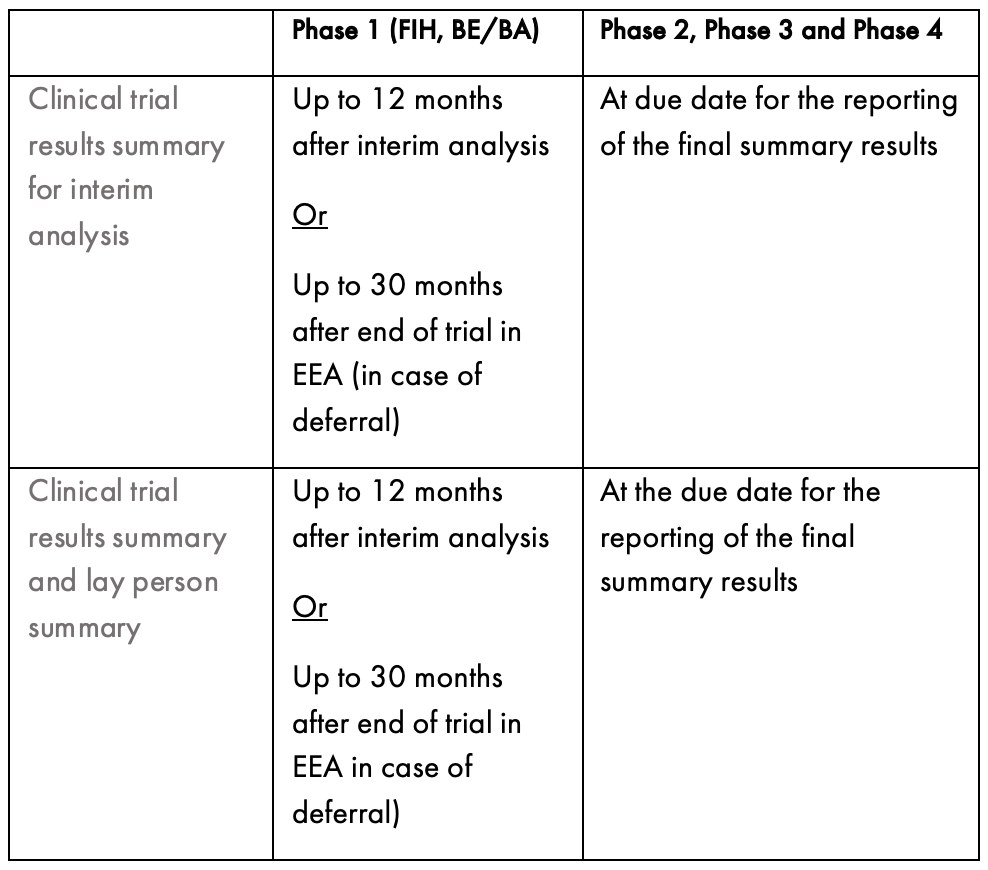
How Sponsors Should Prepare to Conduct Clinical Trials in the EU.
The CTR is aimed at facilitating interactions with the regulatory authorities of the EU Member States.
However, as described above, it requires the use of a new submission portal, a decision regarding how to approach Part I and Part II submissions (in parallel or separately) and familiarisation with several new requirements.
From the 31 January 2023 all CTA applications for trials to be conducted in the EU/EEA must be compliant with the CTR.
In order to be ready to conduct trials under the CTR preparation is needed. If sponsors do not delegate all of the tasks for conducting a clinical trial to a CRO, they have to adapt their standard operation procedures in order to be ready to meet the new requirements regarding preparation of trial documents, application and maintenance procedures. In addition, such sponsors need to be able to complete both the Part I and Part II submission steps. The timeframe to reply to requests for further information is limited. Sponsors should define internal procedures to receive RFIs in CTIS and to respond promptly. This might involve a re-distribution of decision rights. Sponsors also need to be aware of the new transparency rules as well as the fact that parallel submission of substantial modifications is no longer allowed.
In conclusion, the CTR looks to further improve on the CT Directive, but all concerned parties - sponsors, regulators, CROs, etc - will need to allow time to learn how to operate under this new legal framework.
Authored in collaboration with FGK.